本文介绍GATK call SNP的原理。
1. 一些概念
- SNP
- DNA序列中单个核苷酸的替代导致的、且分布于种群中相当一部分个体(如:1%以上)中的基因多样性。
- SNP具有二态性,几乎所有常见的单核苷酸多态性(SNP)位点只有两个等位基因。
- 单核苷酸态性(SNP)位点的分布是不均匀的,在非编码区比在编码区更常见。这是由于自然选择倾向于保留最益于遗传适应性的单核苷酸多态性(SNP)位点。
- 转换的发生率总是明显高于颠换。其中,C→T概率总是更高,这是因为CpG二核苷酸上的胞嘧啶残基大多数是甲基化的,可自发地脱去氨基而形成胸腺嘧啶。
- SNP与突变
- SNP是DNA序列中常见的单个碱基变异,而突变是DNA序列中更广泛的、相对罕见的变异。DNA序列有些地方,人和人就是不一样,这种不一样就叫多态性(polymorphism),单个碱基的不同,就叫SNP。如果这个SNP是突变引起的,依然可以叫它点突变。
- SNP与SNV
- SNP是SNV的一种特殊情况,SNV(Single Nucleotide Variant)则是指单个碱基的变异,包括SNP在内。
- SNV范围更广,它还包括其他类型的单个碱基变异,例如单个碱基插入或缺失等indel。
- 群体中某个单碱基变异的频率达到一定水平就叫SNP,而频率未知(比如仅仅在一个个体中发现)就叫SNV。
- SNP calling与genotype calling
- SNP calling指的是在基因组序列数据中识别SNP的过程。SNP calling只是确定那个基因组位点存在变异,并不涉及对应位点的基因型。
- genotype calling则是在识别出SNP后,进一步确定每个样本的基因型的过程。例如是纯和的还是杂和的等等。这个过程通常会根据每个样本中的SNP位点的碱基信息,来决定该样本在该位点上的基因型,例如AA、AT、TT等。
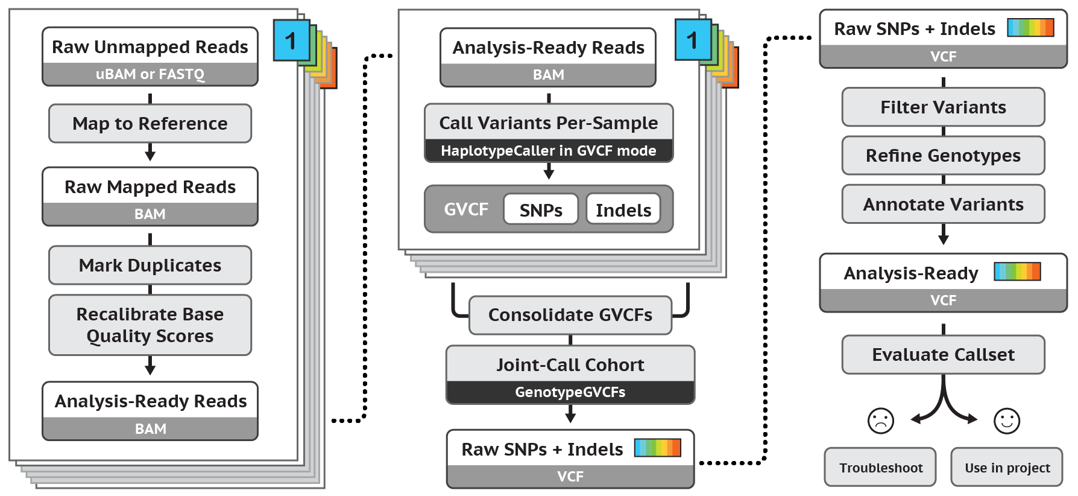
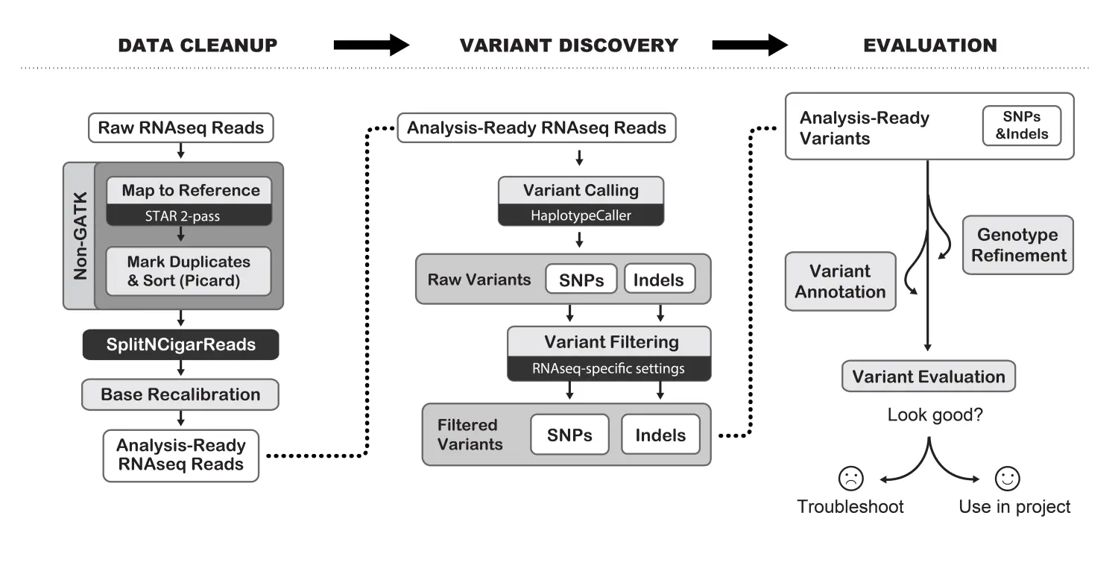
2. DNA-seq与RNA-seq数据call SNP原理
DNA-seq call SNP步骤:
RNA-seq call SNP步骤:
关键步骤:
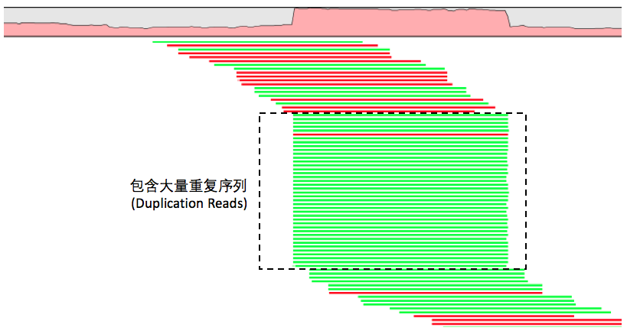
去除PCR重复(for DNA-seq and RNA-seq)
- PCR重复会影响SNP位点的可信度,DNA在打断的那一步会发生一些损失,主要表现是会引发一些碱基发生颠换变换(嘌呤-变嘧啶或者嘧啶变嘌呤),带来假的变异。PCR过程会扩大这个信号,导致最后的检测结果中混入了假的结果;
- PCR反应过程中也会带来新的碱基错误。发生在前几轮的PCR扩增发生的错误会在后续的PCR过程中扩大,同样带来假的变异;
- 对于真实的变异,PCR反应可能会对包含某一个碱基的DNA模版扩增更加剧烈(这个现象称为PCR Bias)。因此,如果反应体系是对含有reference allele的模板扩增偏向强烈,那么变异碱基的信息会变小,从而会导致假阴。
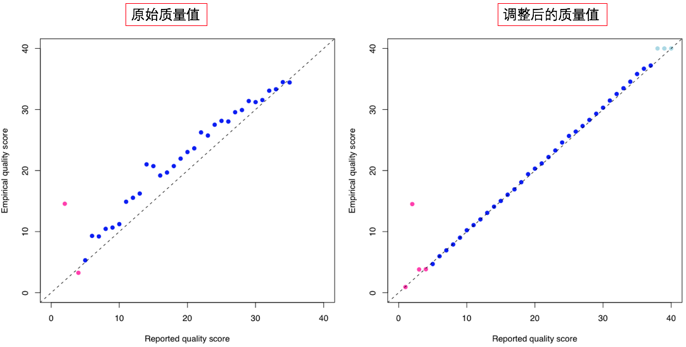
BQSR 碱基质量校正:
- BQSR 根据已知的SNP位点,基于机器学习的方法进行碱基质量校正,校正测序错误可能造成的误差,只会调整非已知SNP区域。
- 对于非人物种,可以先call出一些SNP进行严格的过滤,然后作为已知的SNP去迭代校正
SplitNCigar (only for RNA-seq)
- 转录信息比对到基因组上时,比对到内含子时为N,SplitNCigarReads可以留下跨越N区域的reads RNA-seq数据还需要去掉内含子区域的reads,对比对中的N进行剪切。
3. SNP calling策略
一般有三种策略,一般推荐第三种策略:
- Single sample calling
- 每一个sample的bam file都进行单独的snp calling,然后每个sample单独snp calling结果再合成一个总的snp calling的结果。
- Batch sample calling
- 一定数目群集的BAM files 分开批量call snps,然后不同批之间再merge在一起
- Joint sample calling
- 所有samples的BAM files一起call 出一个包含所有samples 变异信息的output
batch calling: sample BAMs are analyzed in separate batches, and batch call sets are merged in a downstream processing step;
classic joint calling: variants are called simultaneously across all sample BAMs, generating a single call set for the entire cohort.
第三种策略对于低频率的变异具有更高更好的检测sensitivity,如图: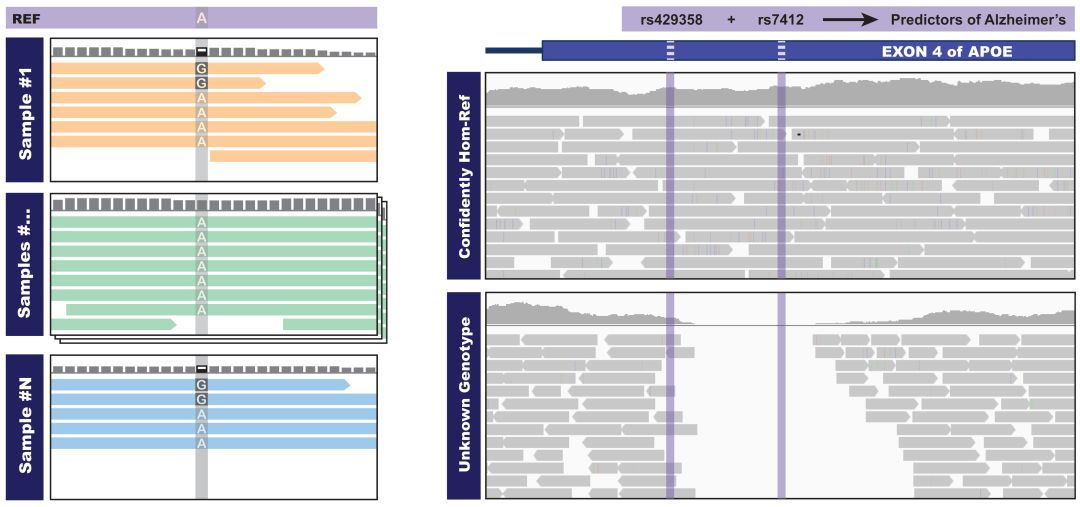
所有samples中所有的位点都是同时call的,换句话说就是,所有位点的信息都是share的,因此可以如果某些samples中个别位点是低频率的,但是可能在其他samples中,该位点的频率比较高,因此可以准确的对低频位点有更加好的calling 效果。
在1到n的samples中,碱基G只出现在其中两个samples中。如果我们将这些samples单独的call snps,这个低频率G的位点将会被忽略。但是joint calling 却可以允许将这些碱基G出现的频率进行累加,将该低频率的突变也call出来。
右图中,上面的sample是一个跟ref一样具有纯合位点,下面的sample在一些位点中有部分数据缺少的情况,例如rs429358.如果将这两个samples分开call snp,然后merge一起,这样会错误地将这两个samples,在这个位点上看作具有类似的变异频率,但是其实由于下面的samples在该位点的区域存在数据缺少,只能看作non-informative。
4. 变异检测原理
HaplotypeCaller通过对活跃区域(也就是与参考基因组不同处较多的区域)局部重组装,同时寻找SNP和INDEL。
当HC看到一个地方好活跃,就不管之前的比对结果,直接对这个地方进行重新组装。
先推断群体的单倍体组合情况,计算各个组合的几率,然后根据这些信息再反推每个样本的基因型组合。
具体步骤如图: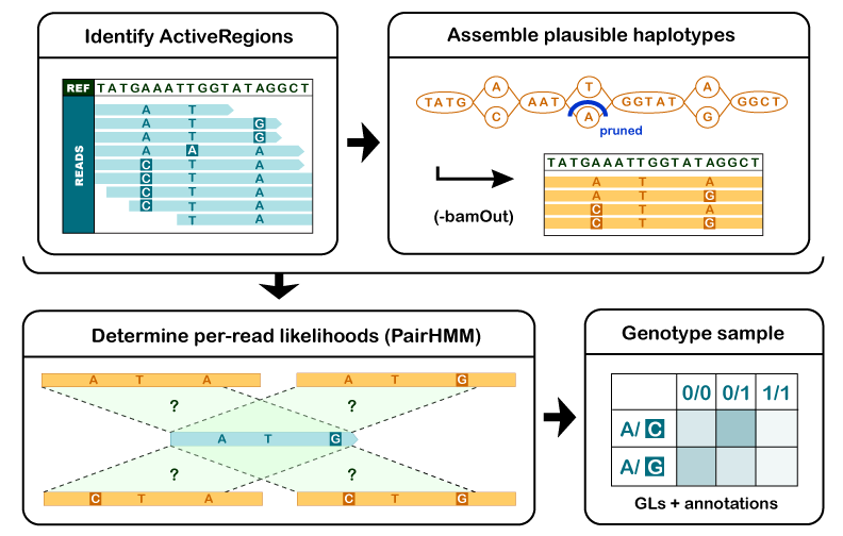
- 从原始测序数据中提取kmer片段,并构建一棵kmer树,以便在后续的变异检测中快速地比对到参考基因组上。
- 对每个kmer片段,使用一种称为”De Bruijn graph”的方法来构建片段的序列图。这个序列图可以将kmer片段映射到参考基因组上,并在变异检测中帮助识别SNP和INDEL等变异。
- 利用序列图对每个样本进行局部基因型估计,即对每个kmer片段,估计该样本中可能存在的变异类型(例如SNP或INDEL)及其频率。这个过程中考虑了对错配比对、reads深度、比对质量等多个因素,以提高估计的准确性。
- 将各个kmer片段的局部基因型拼接起来,得到样本的全局基因型估计,并进行更准确的变异检测。在这个过程中,HaplotypeCaller会考虑到不同片段之间的连接关系,以提高变异检测的准确性。
- 进行变异过滤,去除可能的假阳性变异,并提取出高置信度的SNP和INDEL等变异。
5. SNP过滤
SNP过滤有两种方式:
- 通过质量校正来过滤 Variant Quality Score Recalibration, VQSR
- 用机器学习的方法基于已知的变异位点对caller给出的原始 variant quality score 进行校正 (VQSR)。需要大量高质量的已知variants作为训练集,一般用于DNA-seq数据。
- 直接过滤 Hard filtering
- RNA-seq进行SNP过滤推荐直接过滤,根据一些特征值进行过滤,如测序深度等。
- GATK推荐的过滤标准:
- QD >=2
- FS <=60
- SOR <=3
- MQ >=40
- MQRankSum >=-2.5
- ReadPosRankSum >=-8
SNP过滤VSQR原理:
- 通过质量校正来过滤 Variant Quality Score Recalibration, VQSR需要一个精心准备的已知变异集,它将作为训练质控模型的真集。GATK会选择数据库里的变异数据作为高质量的已知变异集,把它们作为VQSR时的真集位点。VQSR是用我们自己的变异数据来训练模型。这些已知变异集的意义是告诉我们群体中哪些位点存在着变异,如果在其他人的数据里能观察到落入这个集合中的变异位点,那么这些被已知集包括的变异就有很大的可能是正确的。也就是说,我们可以从数据中筛选出那些和真集『位点』相同的变异,把它们当作是真实的变异结果。接着,进行VQSR的时候,程序就可以用这个筛选出来的数据作为真集数据来训练,并构造模型了。
- VQSR的核心就在于构造一个区分“好”变异和“坏”变异的分类器。这个分类器通过GMM模型构造,在构造的时候并不是用所有数据来造的。而是挑出部分和已知的不同变异集合Overlap的位点 (如与千人数据集、Hapmap数据集等)一并分配不同的高可信度权重,基于群体遗传的原理,已知的变异会被认为是更加靠谱的变异,因此在初始化的时候先把它们当作是好也就是正确的变异。
- 这个初始变异集很重要,然后利用这些好变异的特征训练一个区分好变异的GMM,之后对全部数据进行打分,再把评分最低的那些拿出来,构成一个最不像正确变异的集合,用来构造一个区分坏变异的GMM,用来专门识别坏变异。最后同时使用好和坏的GMM再一次同时对变异进行打分,看每个变异更像谁,就能够评判出这个变异可信的质量值了!越靠近好的GMM,质量自然就越高。这就是VQSR过滤的大致原理了。
参考资料: