本文介绍了Ribo-seq的技术原理和上游的数据处理。
- 技术原理
- 数据质控
- 比对
1. Ribo-seq技术原理
Ribo-seq建库测序的原理主要是通过对细胞使用翻译抑制剂CHX,使正在翻译的核糖体固定在mRNA序列或者起始位点上。裂解细胞后在细胞裂解液中加入RNase,正在翻译的mRNA片段由于核糖体的保护而保留下来,消化不受核糖体保护的mRNA,分离单一核糖体并提取纯化核糖体上未被消化的短片段mRNA,进行建库测序和相应的数据分析。Ribo-seq测序得到的片段大小在28-32nt左右。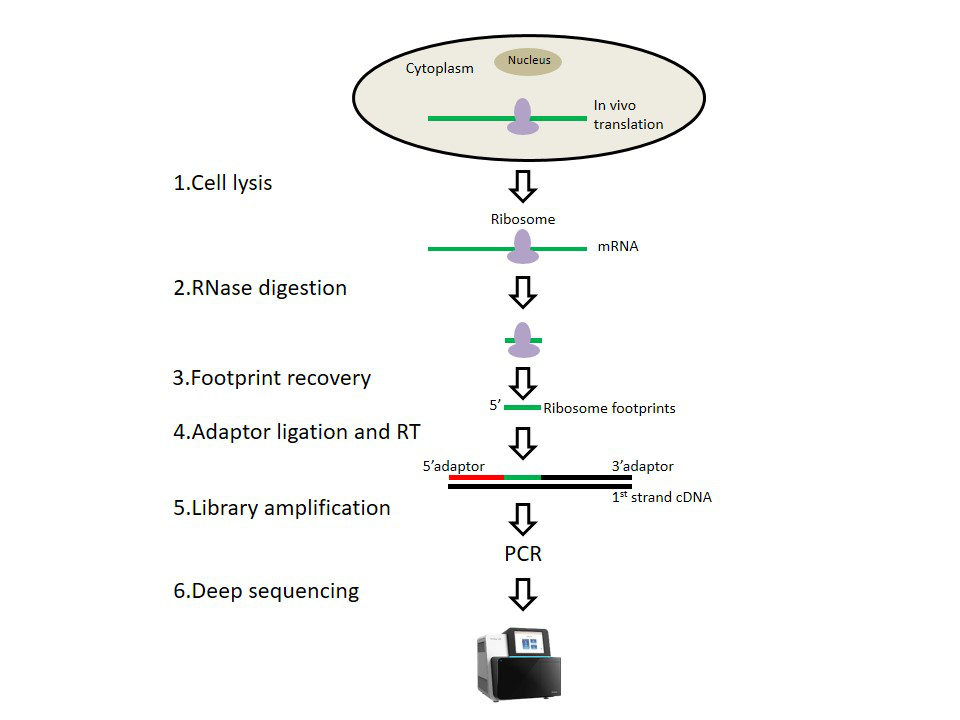
2. Ribo-seq数据质控
Ribo-seq的下机数据处理部分和RNA-seq的基本流程是一样的,但是Ribo-seq多了去除rRNA这一步,RNA-seq一般情况下是不需要去除rRNA,但是Ribo-seq测序的结果中会有很大含量的rRNA,如果不去除,会对后续的结果造成很大影响,必须要去除。
2.1 去接头
trim_galore软件封装了fastqc和cutadapt,可以进行质控也可以去接头,如果不知道接头序列,也可以不指定序列,软件可以检测接头序列(但是我试了一下自动检测没有成功,检测不到)。
参数解释:
-j:线程数。
–quality/-q:设定Phred quality score阈值,默认为20。
–phred33::选择-phred33或者-phred64,表示测序平台使用的Phred quality score。
–adapter/-a:输入adapter序列。也可以不输入,Trim Galore!会自动寻找可能性最高的平台对应的adapter。自动搜选的平台三个,也直接显式输入这三种平台,即–illumina、–nextera和–small_rna。
–stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1(非常苛刻)。可以适度放宽,因为后一个adapter几乎不可能被测序仪读到。
–length:设定输出reads长度阈值,小于设定值会被抛弃。
–paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准。
–gzip和–dont_gzip:清洗后的数据gzip压缩或者不压缩。
–fastqc:trim之后,自动进行fastqc质控。
单端测序:
1 | for i in `cat acc_list.txt` |
双端测序:
1 | for i in `cat acc_list.txt` |
2.2 去除rRNA和tRNA
核糖体保护的片段很短,因此在提取过程中会有许多其他序列的污染,rRNA和tRNA是细胞中占比很大的部分,因此拿到测序数据后需要将数据比对到rRNA/tRNA上去除这些无用的数据。在比对的时候,我们可以不用选择很严格的标准,我们的目的是为了尽可能多的去除rRNA,因此我们可以使用bowtie2的局部比对,只要一部分片段比对上,我们就认为是rRNA或tRNA,从而将其去除。
首先,建立索引:
1 | bowtie2-build 02.remove_rRNA_tRNA/rRNA.fasta 02.remove_rRNA_tRNA/rRNA-index/rRNA |
然后进行比对:
参数解释:
–local:用局部比对
–end-to-end:全局比对
-x:索引文件所在位置的名字前缀
-p:线程数
-U:单端测序
–un:单端测序比对不上的片段输出
-S:比对情况输出sam
–un-conc:双端测序中比对不上的片段
单端测序比对命令:
1 | for i in `cat acc_list.txt` |
双端测序比对命令:
1 | for i in `cat acc_list.txt` |
3 Ribo-seq比对
去除rRNA、tRNA之后,就可以进行比对了。由于Ribo-seq测序得到的是正在翻译的片段,就表明这些序列是具有编码能力的mRNA,因此,比对到全基因组上一般是不准确的,所以一般要比对到转录本序列,一般选择CDS最长的转录本序列进行比对。
3.1 比对
由于Ribo-seq测序得到的序列比较短,因此我们可以选择bowtie进行比对。
参数解释:
-v模式参数:-v V其中V为整数。-v V代表全长错配不能超过V个,可以是0-3。这时,不考虑是否高保真区,也不考虑Phred quality值。
–best与–strata
–best参数代表报告文件中,每个短序列的匹配结果将按匹配质量由高到低排序。–strata参数必须与–best参数一起使用,其作用是只报告质量最高的那部分。所谓质量高低,其实就是指错配的碱基数,如果指定了-l L参数,那就是在高保真区内的错配数,否则就是全序列的错配数。如果你还指定了 -M X的话,那就会在质量最高的当中,随机选择X个来报告。也就是说,当我们指定了-M 1 –best –strata的话,那就只报告1个最好的。
-m:指定同一个序列允许最多比对到基因组上几次。
这里用的参数:最多允许两次错配,一个序列只能在基因组上比对一次。
1 | bowtie-build 05.transcript_index/longest_cds_trans.fa 05.transcript_index/longest_trans |
3.2 定量
htseq-count进行定量,bam文件中包含了比对上的reads和没有比对上的reads, 只有比对上的reads 会用来计数,htseq-count默认会根据mapping的质量值对BAM文件进行过滤,默认值为10, 意味着只有mapping quality > 10的reads才会用来计数,当然可以通过-a参数来修改这个阈值。
。
参数解释:
-f:输入文件格式,
-s:链特异性
-i:设置feature ID是由gtf/gff文件第9列那个标签决定的;若gtf/gff文件多行具有相同的feature ID,则它们来自同一个feature,程序会计算这些features的表达量之和赋给相应的feature ID。
-t:程序会对该指定的feature(gtf/gff文件第三列)进行表达量计算,而gtf/gff文件中其它的feature都会被忽略,一般为exon/CDS。
-m:设置表达量计算模式。该参数的值可以有union, intersection-strict and intersection-nonempty。这三种模式的选择请见上面对这3种模式的示意图。从图中可知,对于原核生物,推荐使用intersection-strict模式;对于真核生物,推荐使用union模式。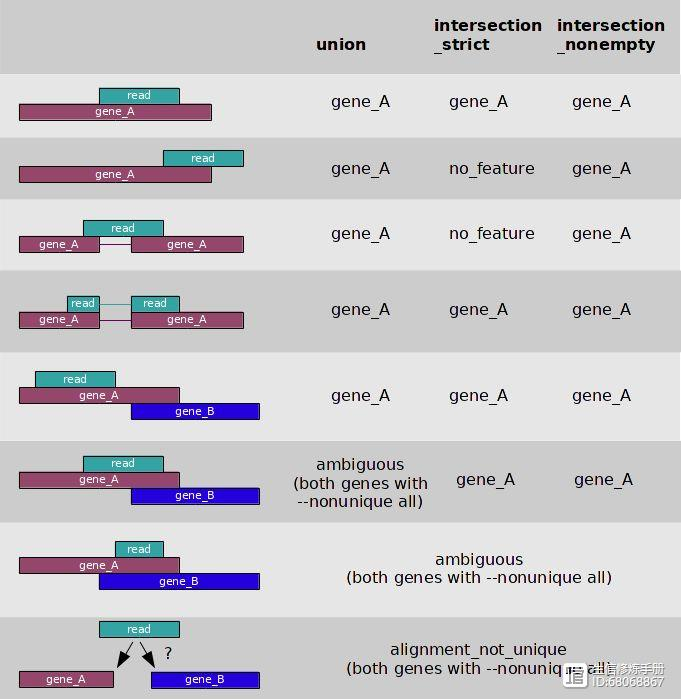
1 | for i in `cat acc_list.txt` |