本文介绍三代hifi基因组测序的组装。
1.HiFi测序
HiFi reads(High fidelity reads)是Sequel II三代测序平台推出的兼顾长读长和高准确度的测序序列,2019年由PacBio公司推出,一般采用CCS(Circular Consensus Sequencing)模式测序,HiFi测序能够产出既有较长读长(10-20kb)又具有高序列精度的测序结果(约99.9%)。
环化共有序列CCS的测序方式:构建一个环形的DNA文库,在这种测序模式下,酶读长一般大于插入片段(即被测序片段)长度,因此酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中造成的随机测序错误,可以通过算法整合和自我纠错校正来去除,最终得到高准确度的HiFi reads。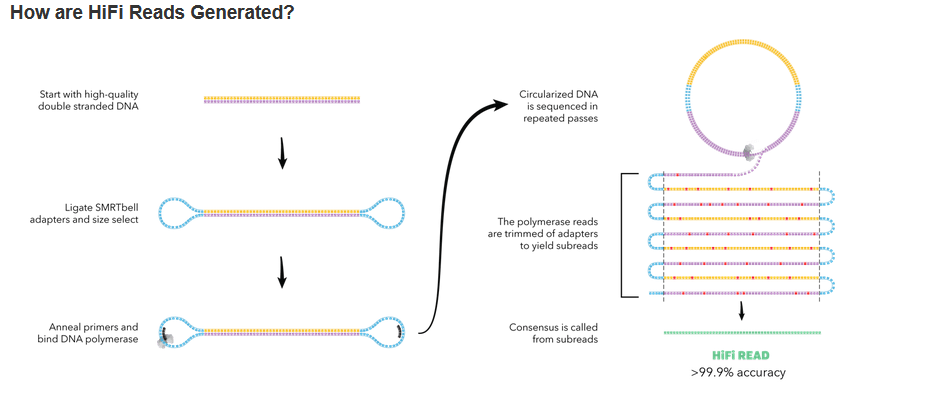
适用于HiFi测序组装的软件有Flye、HiCanu(是Canu软件专门针对HiFi reads优化版本)、hifiasm(速度快、专注解析单倍型)。
2.HiFi测序下机数据
Sequal平台的下机数据主要有三种:bam文件、bam.pbi文件、xml文件。
2.1 BAM文件
Pacbio下机数据中的BAM文件是没有比对过的,用于储存序列。
第一列:reads信息
{movieName}/{holeNumber}/{qStart}_{qEnd}
(对于CCS:{movieName}/{holeNumber}/ccs)
MovieName 是cell的名字,holeNumer是ZMW孔的编号,qStart和qEnd是subreads相对于ZMW reads的位置。
第二列 (sum of flags):比对信息,均为4 代表没有比对上,也表明了bam文件只储存了序列信息,而没有比对信息。
第三列 (RNAM):参考序列,值为*,代表无参考序列
第四列 (position) : 比对上的第一个碱基位置 0
第五列 (Mapping quality) : 比对质量分数 255
第六列 (CIGAR值) : 比对的具体情况
第七列 (MRNM, ) : mate 对应的染色体
第八列 (mate position) : mate对应的位置 0
第九列 (ISIZE, Inferred fragment size) : 推断的插入片段大小 0
第十列 (Sequence) : 序列信息 具体的ATCG
第十一列 (ASCII码) : 碱基质量分数 ASCII+33
第十二列 : 可选区域记录Reads的总体属性包括信号长度,信号强度等信息。
2.2 bam.pbi
BAM文件的索引文件。
3.hifi reads组装
3.1 flye组装
用flye进行组装,输入是测序的fasta或fastq文件,所以需要将下机bam文件转换为fasta文件。bam2fastx软件可以实现转换,也可使用samtools转换。
1 | # bam to fasta |
完成转换之后,可以用flye进行组装。
1 | # assembly |
组装结果,输出文件:
assembly.fasta:最终组装结果
assembly_graph.gfa/assembly_graph.gv:
assembly_info.txt:
3.2.hifiasm组装
hifiasm擅长组装单倍型,其组装有三种模式:只有hifi reads、Hifi reads+ Hi-C、hifi reads+双亲二代测序的Trio-binning。
hifiasm安装
1 | #install |
hifiasm组装,输出为assembly.fa
1 | # 只有hifi reads |
4. quast评估
完成基因组组装之后,要用quast评估组装结果,quast主要是计算contigs数量、总长度、N50、N90、L50、GC rate等等,可以根据参考基因组进行评估,也可以不依据参考基因组进行评估。软件使用:
1 | #install |
评估指标:
1.contig size
No. of contigs: 组装中的 contigs 总个数。
Largest contig: 最长 contig 的长度。
Total length: assembly 中的碱基总数。
Nx (where 0<=x<=100): 最长长度的 contigs 占组装碱基的百分比。
NGx, Genome Nx: 相等或更长的重叠群产生参考基因组长度的 x% 的重叠群长度,而不是组装结果长度的 x%。
2.基因组相关统计
Genome fraction (%) : 基因组中被组装结果覆盖到的碱基数 除以 参考序列的总长度得到的比值;位于重复区域的contig可能会比对到多个位置,因此会被重复计算。
Duplication ratio:组装结果中可比对到基因组的碱基数 与 基因组中被覆盖到的碱基数的比值;如果组装的结果中重复序列较多,多个contig覆盖同一个基因组区域的话,这个值会大于1。这种情况可能是由于过多的估计了重复序列的拷贝数。
Largest alignment:将组装结果同基因组进行比对,得到的最大的连续的比对的长度;这个值一般会同largest contig相同,但是如果larget contig有些missassembled,或者部分未比对上去的话,会相对小一些。
Total aligned length:组装结果中可比对到基因组的碱基数,由于存在未比对和部分未比对,这个数字一般会小于组装的结果的总长度(total length)。
3.组装错误等,这些只能根据已知参考基因组进行评估。
No. of misassemblies: 组装错误的数量。
No. of misassembled contigs: 包含错误组装断点的 contigs 数量。
Misassembled contigs length: 在所有 contig 中包含一个或多个错配的碱基总数。
No. of unaligned contigs: 与参考序列没有对齐的 contigs 数量。
No. of ambiguously mapped contigs: 在参考基因组的多个位置具有相同质量的高得分参考比对的 contigs 数量。
除了以上汇总统计数据之外,Quast 还生成包含每个 contig 详细信息的报告,包括 contig 是否未对齐、不明群映射、错误组装或正确。
4.未比对上的。
fully unaligned contigs:没有比对上参考基因组的contig数
Fully unaligned length:没有比对上参考基因组的contig总长度
partially unaligned contigs:部分没有比对上基因组的contig数,长度大于参数值(500bp)
Partially unaligned length:部分没有比对上基因组的contig中总的没有比对上的碱基数
5.错配的
mismatches :所有比对上的碱基中,mismatch的个数
indels :所有比对上的碱基中,indel的个数
Indels length :indel的总长度
mismatches per 100 kbp:每100kbp中,mismatch的个数
indels per 100 kbp : 每100kbp中,indel的个数
indels (<= 5 bp) : 长度小于5的indel个数
indels (> 5 bp) : 长度大于5bp的indel个数
N’s :N碱基的个数
N’s per 100 kbp:每100kbp中N碱基的个数
5.进行polish
三代测序读长长,但是也有较高的错误率,组装完成后可以用pilon进行polish,可以用二代测序(pilon)或三代数据(gcpp或racon)再次纠错。这里,介绍gcpp,gcpp封装在pb-assembly中,可以通过conda安装pb-assembly,也可以直接安装gcpp。
1 | # use NGS pair end data or hifi reads to polish, here, we use hifi reads to polish hifi-assembly |
6.busco评估基因组组装完整性
conda install -c bioconda busco
待更新