- SeuratObject数据结构
- Assays
- meta.data
- active.assay
- active.ident
- reduction
- 提取数据
- 提取基因ID和细胞ID
- 提取细胞坐标信息
- 提取表达矩阵
- 提取平均表达量
- 聚类细胞数目与比例统计
- 修改聚类后的idents
- 提取部分Seurat对象
1. SeuratObject数据结构
首先,先来了解一下SeuratObject的数据结构和格式。
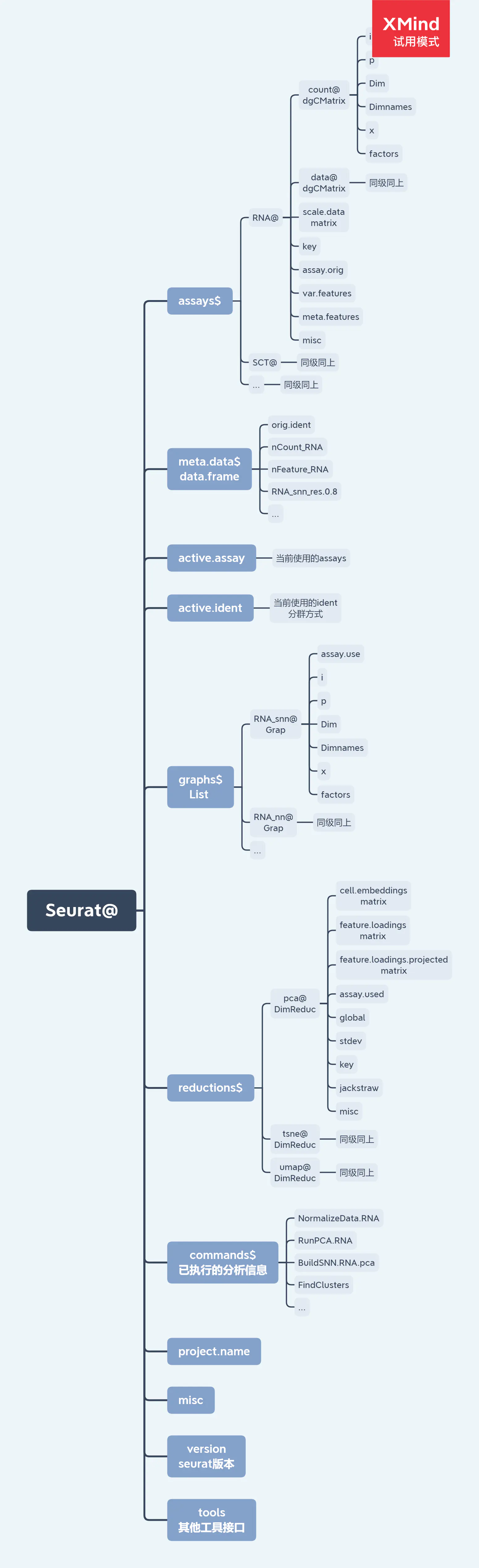
图片来源:https://www.jianshu.com/p/13142bf51e81
1.1 Assays
默认情况下Seurat对象的Assay是”RNA”,在后续的数据处理过程中,会产生新的。例如,integration之后、SCTransform之后等过程处理之后,都会产生新的Assay,存放的是经过处理之后产生的矩阵。修改默认的Assay可以通过DefaultAssay函数,也可通过 @active.assay查看默认的assay。
1
2
| pbmc@active.assay
DefaultAssay(pbmc) <- "RNA"
|
在assay数据中,counts是原始数据,data是normalize(归一化)之后的数据,scale.data是scale(标准化)之后的数据。var.features是高变的features,默认是2000个。调用方法:
1
2
| pbmc@assays$RNA@counts
pbmc[["RNA"]]@counts
|
meta.data数据中储存的是细胞的相关信息,主要包括:orig.ident,nCount_RNA,nFeature_RNA,percent.mito,RNA_snn_res.0.4,seurat_clusters等。orig.ident是细胞的来源信息,例如我的是3个样本合并之后的dataset,那么这个里面就是sample1、sample2、sample3,nCount_RNA是每个细胞中RNA数量,nFeature_RNA同理,percent.mito每个细胞中的线粒体基因占比信息,seurat_clusters是细胞聚类之后被归为哪一类。调用方法:
1
2
| pbmc$orig.ident
pbmc[["orig.ident"]]
|
1.3 active.assay
查看当前的默认assay。调用方式:
1.4 active.ident
查看使用的分群方式。调用方式:
1.5 reduction
储存降维之后每个细胞的坐标信息,3种方法分别在:pca、tsne、umap。坐标信息在cell.embeddings中;每个基因对pca成分的贡献度在feature.loadings中。assay.used指这个降维使用的assay。调用方法:
1
2
| head(pbmc@reduction$umap@cell.embeddings)
head(pbmc@reduction$umap@feature.loadings)
|
2. 提取数据
2.1 提取基因ID与细胞ID
1
2
3
4
5
6
7
8
9
10
11
| rownames(pbmc)
colnames(pbmc)
Cells(pbmc)
WhichCells(pbmc,ident=c(1,2))
WhichCells(pbmc,ident=c(1,2),invert=TRUE)
WhichCells(pbmc,expression=gene1>1,slot="counts")
|
2.2 提取细胞坐标信息
1
| Embeddings(pbmc,reduction="umap")
|
2.3 提取表达矩阵
1
2
3
4
5
6
|
raw <- as.matrix(GetAssayData(pbmc,slot="counts"))
raw <- as.matrix(pbmc@assays$RNA@counts)
raw5 <- as.matrix(GetAssayData(pbmc,slot="counts")[,WhichCells(pbmc,ident="5")])
|
2.4 提取平均表达量
1
2
| cluster.averages <- AverageExpression(pbmc)
write.table(cluster.averages[["RNA"]][,1:15],"cluster.averages.txt")
|
2.5 聚类细胞数目与比例统计
1
2
3
4
5
6
7
8
9
10
11
12
13
|
table(Idents(pbmc))
table(pbmc$RNA_snn_res.0.4)
table(pbmc$orig.ident)
table(Idents(pbmc),pbmc$orig.ident)
prop.table(table(Idents(pbmc)))
prop.table(table(pbmc$RNA_snn_res.0.4))
|
2.6 修改聚类后的idents
1
| pbmc <- RenameIdents(pbmc,`0`="B cells",`1`="NK cells")
|
2.7 提取部分Seurat对象
1
2
3
4
5
6
|
subset(pbmc,cells=cells)
subset(pbmc,ident=c(1,2,3))
subset(pbmc,ident=c(1,2,3),invert=TRUE)
|
参考
1.https://www.jianshu.com/p/13142bf51e81
2.https://www.jianshu.com/p/1db7c28249d4