本文介绍从fastq文件开始,用cellranger进行定量:
- 用cellranger构建参考基因组
- cellranger mkgtf
- cellranger mkref
- cellranger count
1. 生成cellranger参考基因组
cellranger官方提供有小鼠和人类的参考基因组,其他物种的需要用mkref自己产生。
1.1 生成用于产生参考基因组的gtf文件
–attribute参数用于指定gtf文件中的biotype。
1 | cellranger mkgtf Dmel.gtf Dmel.BDGP6.32.104.filtered.gtf \ |
1.2 生成参考基因组文件
接下来根据上一步产生的gtf文件生成参考基因组文件,–genome参数指定接下来产生的参考基因组的文件夹名字,–fasta为下载的参考基因组的序列文件,–gtf为上一步产生的gtf文件。
1 | cellranger mkref --genome=Dmel.genome \ |
2. cellranger count定量
有了fastq和参考基因组,接下来就可以定量了。
–id输出文件夹名字,–transcriptome参考基因组所在的文件夹,–fastqs为fastq序列所在的文件名,–sample用于分析的文件prefix,–localcores线程数,–localmem内存数,加上–nosecondary参数不进行下游聚类分析。
1 | cellranger count --id=run_sample1 \ |
定量结束之后,产生的文件夹里outs/下会有一个网页文件web_summary.html,打开会看到下图的信息。
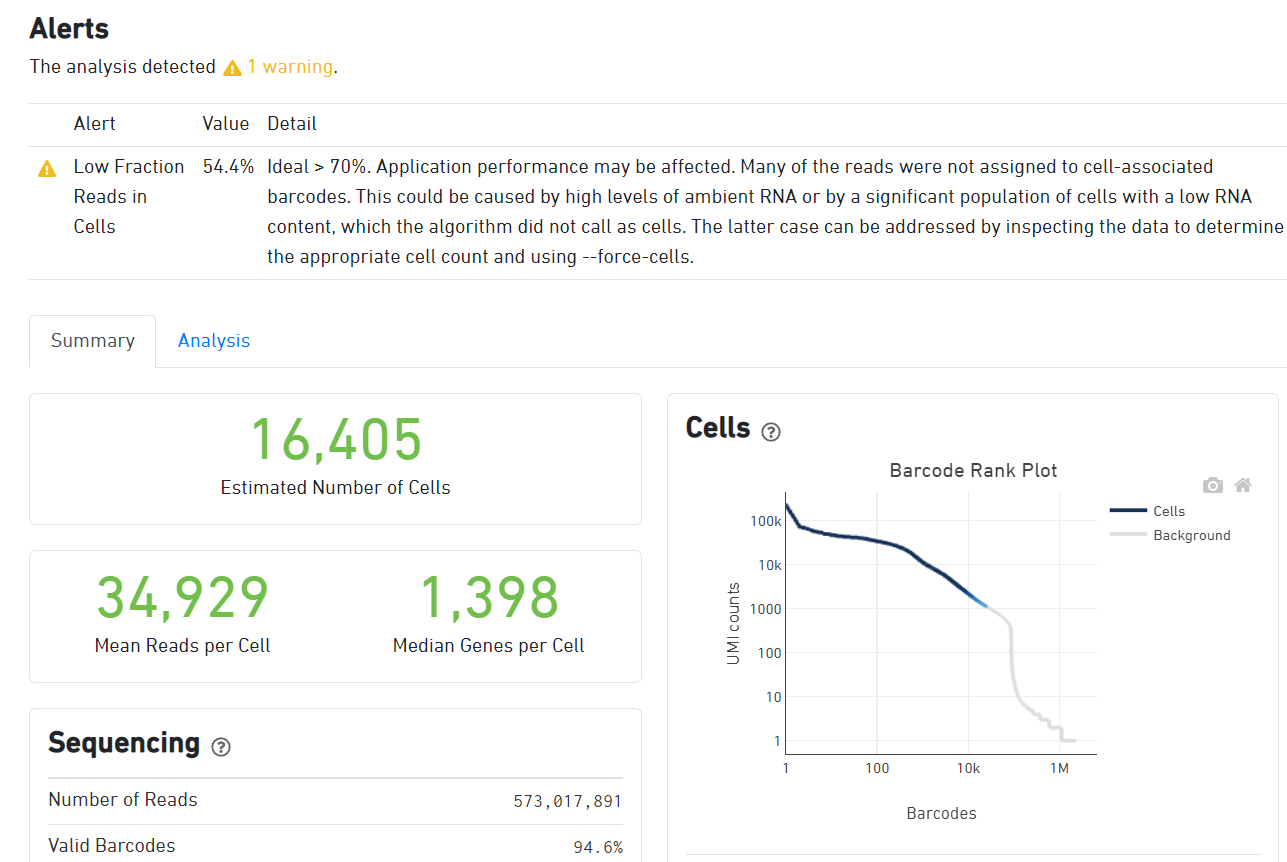
发现有个warning:Low fraction reads in cells,显示我的数据大概在54%左右,理想情况下应该为70%,官方的解释有两种可能:1.环境RNA水平过高;2.有一部分细胞RNA content很低,若是第二种情况,那么可以在count步骤加上–force-cells参数,设置一下自己预期的细胞数量,从而去掉RNA含量低的细胞。但是我设置了这个参数之后,还是会有这个warning,这可能说明细胞中reads比例低不是第二种情况造成的。
1 | cellranger count --id=run_sample1 \ |